Generality
Cystic fibrosis is the most common autosomal recessive disease in the Caucasian population, affecting approximately 1 individual in every 2, 500.
This pathological condition is known for its harmful effects on the respiratory system, but it also affects other systems such as the digestive and reproductive systems.
In individuals with cystic fibrosis, the airways are clogged with thick, viscous mucus, which is difficult to remove even with the most energetic cough. Breathing becomes difficult and patients - if continuous efforts are not made to keep the airways clean several times a day - risk dying due to their own secretion. Cystic fibrosis patients die frequently due to pneumonia, as the clogged airways provide a fertile environment for bacterial growth.

Causes
Cystic fibrosis is caused by mutations in cystic fibrosis fibrosis (CFTR) transmembrane conductance gene localized at chromosome 7 (locus mapping: 7q31).
At least 1, 500 mutations of the CFTR gene are known. The most frequent mutation is commonly called "Delta-F508" (DF508) and is determined by the deletion of 3 base pairs in exon 10, which results in the loss of phenylalanine at position 508.
The protein encoded by the CFTR gene is a transmembrane channel belonging to the ATPase superfamily of trafficking or ABC transporters, located at the level of the apical membrane of epithelial cells and deputed to the transport of chlorine ion.
Under normal conditions, particular cells that line the airways secrete mucus together with an aqueous liquid that decreases their density. In cystic fibrosis the secretion of the aqueous fluid is greatly reduced , consequently the mucus becomes very dense and difficult to remove from the respiratory tract.
In the respiratory epithelium, like all the epithelia that carry liquids, the transport of water depends on the transport of solutes. To secrete water, the cells of the respiratory epithelium actively transport chlorine ions (Cl-) from the interstitial fluid to the lumen, creating a negative electric potential that causes a passive flow of sodium (Na +) in the same direction. The movements of Na + and Cl- raise the osmotic pressure of the liquid that wets the slope of the epithelium turned towards the lumen, consequently the water moves passively according to the osmotic gradient, from the interstitial liquid to the lumen. The gene defect that affects the onset of cystic fibrosis directly prevents the transport of Cl- and indirectly interferes with the transport of Na + and water. Consequently the osmotic gradient necessary for the secretion of water is not created in the epithelium.
Risk factors
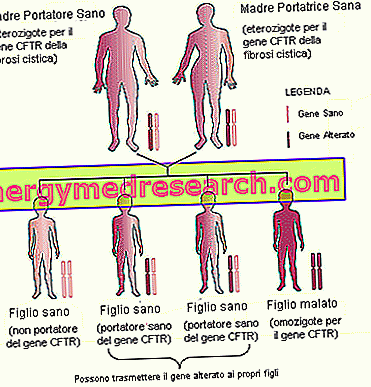
- Family inheritance . Considering that cystic fibrosis is a hereditary disease, which is transmitted in an autosomal recessive manner, it is important to consider the family history (anamnesis) of future parents.
- Breed . The incidence of cystic fibrosis is greater in white people in the north and of European origin.

Symptoms and Clinical Signs
To learn more: Cystic Fibrosis Symptoms
The severity of symptoms can vary, depending on the course of the disease: most clinical signs influence the respiratory system and the gastro-intestinal system.
Respiratory signs and symptoms
The thick and viscous mucus associated with cystic fibrosis causes airway obstruction and inhibits the activity of the cilia that line the pulmonary epithelium, facilitating the elimination of the mucus itself. In the small airways, the resulting mucous stagnation contributes to creating an ideal environment for bacteria, such as Streptococcus aureus or Pseudomonas aeruginosa, which predispose the affected subject to recurrent infections and inflammations, which in turn can also be severe, like pneumonia. The chronic nature of these inflammatory processes can damage lung tissue over time and consequently lead to poor respiratory function.
Thus, the characteristic symptoms that develop during the course of cystic fibrosis include:
- Persistent cough with sputum production
- Wheezing breath
- Shortness of breath and difficulty breathing
- Recurrent lung infections
Digestive tract signs and symptoms
Thick mucus can also occlude the ducts that carry digestive enzymes from the pancreas to the small intestine. Without these digestive enzymes, the intestine cannot digest and completely absorb the nutrients in the food taken (in particular, fats, cholesterol and fat-soluble vitamins A, D, E and K).
The result is often the following:
- Swollen abdomen, meteorism, steatorrhea
- Poor weight gain and poor growth (due to malnutrition)
- Intestinal blockage, particularly in newborns (meconium ileum)
- Constipation or severe constipation
Complications
Respiratory system complications
- Bronchiectasia : cystic fibrosis is one of the main causes of bronchiectasia, a condition that damages the airways, resulting in obstructive ventilatory failure, with a marked reduction in expiratory flow.
- Chronic infections : the thick mucus that stagnates in the lungs and sinuses offers an ideal breeding ground for bacteria and fungi. As a result, people with cystic fibrosis may have frequent attacks of sinusitis, bronchitis or pneumonia.
- Nasal polyps : since the inner lining of the nose is inflamed and swollen, soft fleshy growths (polyps) can develop, which can hinder breathing during sleep.
- Cough with blood (hemoptysis): over time, cystic fibrosis can cause thinning of the airway walls. As a consequence, patients with cystic fibrosis can emit blood from the airways, usually through a cough.
- Pneumothorax : this condition, which consists of the accumulation of air in the pleural cavity, is more common in elderly people with cystic fibrosis. Pneumothorax can cause chest pain and shortness of breath.
- Lung collapse : chronic pulmonary infections can damage the lung parenchyma, making lung collapse more likely.
- Respiratory insufficiency : over time, cystic fibrosis can damage lung tissue and compromise its functionality.
Complications of the digestive system
- Nutritional deficiencies : thick mucus can block the pancreatic ducts that carry digestive enzymes from the pancreas to the small intestine. Without these enzymes, the body cannot absorb proteins, fats and fat-soluble vitamins.
- Diabetes : pancreatic beta cells secrete insulin, which is necessary to mediate blood glucose regulation. Cystic fibrosis increases the risk of developing diabetes. About 20 percent of affected people develop diabetes at age 30, so patients are tested for blood glucose at regular intervals.
- Obstruction of the bile ducts : the duct that carries bile from the liver, and from the gall bladder to the small intestine can become blocked and inflamed, leading to liver problems and, sometimes, gallstones.
- Rectal prolapse : frequent coughing or exertion during defecation (constipation) can cause the internal rectal tissue to protrude out of the anus.
- Intussusception : children with cystic fibrosis are at higher risk of intussusception, a condition in which part of the intestinal folds fold back on themselves, creating invaginations. The result is intestinal obstruction, an emergency situation that requires immediate treatment.
Complications of the reproductive system
Almost all men with cystic fibrosis are sterile, because the ducts that connect the testicles and the prostate gland (vas deferens) are blocked by mucus or missing altogether (the absence of the ducts is still a cause unknown).
Although women with cystic fibrosis may be less fertile than other women, they still retain the ability to conceive and bring the pregnancy to an end. However, gestation can contribute to a worsening of cystic fibrosis symptoms.
Other complications
Osteoporosis : people with cystic fibrosis are at a higher risk of developing osteoporosis, with consequent loss of mass and bone strength. Patients should be advised to take calcium, vitamin D and bisphosphonates as appropriate.
Electrolyte imbalances : people with cystic fibrosis tend to have a higher than normal level of salt in sweat. Signs and symptoms such as an increase in heart rate, fatigue, weakness and low blood pressure help to highlight the decompensation that the hydro-saline balance in the blood undergoes.
Diagnosis and Therapy of Cystic Fibrosis »


