Generality
Craniosynostosis is the term by which doctors indicate an abnormality of the skull due to the premature fusion of one or more cranial sutures.
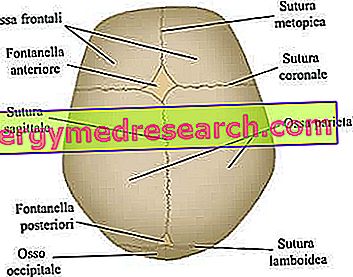
Cranial sutures are the fibrous joints that bind together the bones of the cranial vault (ie the frontal, temporal, parietal and occipital bones).
Craniosynostosis can be an isolated phenomenon (non-syndromic craniosynostosis) or the result of some particular morbid conditions (syndromic craniosynostosis). Among the morbid conditions that cause a premature fusion of the cranial sutures, the best known are: Crouzon syndrome and Apert syndrome.
With the premature fusion of the cranial sutures, the brain structures do not have the appropriate space to grow. This has various consequences, including mainly the increase in intracranial pressure (intracranial hypertension).
A prompt and accurate diagnosis allows to plan an ad hoc treatment. The latter is of a surgical type and has as its final objective the separation of fusesi sutures early.
Recalls of anatomy of the human skull
Equipped with bones and cartilages, the skull is the skeletal structure of the head that constitutes the face and protects the brain, the cerebellum, the brainstem and the sensory organs.

To simplify the study and understanding of the skull, the anatomists have thought of dividing it into two compartments, called neurocranium and splancnocranium .
neurocranium
The neurocranium is the upper cranial region, containing the brain and some of the main sensory organs. Its most important bones - strictly flat - are the frontal, temporal, parietal and occipital bones; these, taken together, form the so-called cranial vault .
splanchnocranium
The splanchocranium, or facial massif, is the antero-inferior region of the skull, composed of even and uneven bones. Represents the skeletal structure of the face, therefore it contains bony elements such as the mandible, upper jaw, cheekbones, nasal bone etc.
What is craniosynostosis?
Craniosynostosis is a rare anomaly of the skull, characterized by an unnatural shape of the head due to the premature fusion of one or more cranial sutures . Cranial sutures are the fibrous joints that bind together the bones of the cranial vault (ie the frontal, temporal, parietal and occipital bones).

From the site: //www.wkomsi.com/
WHEN SHOULD THE CLOSURE OF CRANAL SUTURES APPEAR?
In normal conditions, the fusion of cranial sutures occurs in the post-natal period (NB: some processes even end at the age of 20). This long process of fusion allows the brain to grow and develop properly.
If, as in the case of craniosynostosis, fusion takes place too early - therefore during prenatal, perinatal * or early childhood - the encephalic elements (brain, cerebellum and brainstem) and some sense organs (eyes in particular) undergo an alteration of shape and growth.
Causes
The pathophysiological process that determines craniosynostosis is the premature fusion of cranial sutures .
This process can represent an isolated phenomenon - where by "isolated" we mean that it is not associated with any particular morbid state - or it can be the consequence of some particular syndromes, almost always of genetic nature.
In light of this, doctors have decided to classify craniosynostosis into two categories:
- Non-syndromic craniosynostosis . The term non-syndromic indicates that the cranial anomaly is not associated with any pathology or other physical defect.
- Syndromic craniosynostosis . The term syndromic means that the cranial malformation is the result of a particular syndrome, in most cases of a genetic type.
NON-SINDROMIC CRANIOSINOSTOSIS
Doctors and researchers have not yet established what the causes of non-syndromic craniosynostosis are.
They proposed various hypotheses - including the influence of environmental factors or hormonal-type problems - but none of these theories found confirmation in the experimental results.
Therefore, to understand the precise origin of the anomaly, further studies are needed in this regard.
CRANIOSINOSTOSI SINDROMICA
According to the latest medical research, there are more than 150 different syndromes, all quite rare, capable of causing craniosynostosis.
Among these syndromes, the best known and most common are:
- Crouzon's syndrome . The result of specific mutations in the genes FGFR2 (chromosome 10) and FGFR3 (chromosome 4), this particular morbid condition affects one newborn every 60, 000 and leads to the presence of anomalies exclusively at the level of the head and face.
- Apert's syndrome . It mainly arises due to mutations in the FGFR2 gene (the same as in Crouzon syndrome) and affects one newborn every 100, 000.
Unlike Crouzon syndrome, the genetic alterations of FGFR2 are such that malformations involve not only the skull and the face, but also hands and feet.
- Pfeiffer's syndrome . It arises due to mutations in the "usual" FGFR2 gene and a gene with similar functions, called FGFR1 (chromosome 8). The peculiarity of these mutations is that - in addition to the deformities of the skull and the face - they also determine: syndactyly, brachydactyly and thumbs and large toes (disproportionate to the other fingers).
Pfeiffer syndrome has an incidence of one case per 100, 000 newborns.
- Saethre-Chotzen's syndrome . It is a genetic condition that affects a newborn every 50, 000 or so. Causes various malformations in the skull, face, hands and feet. Some specific mutations of the TWIST1 gene, located on chromosome 7, are responsible for the onset of Saethre-Chotzen syndrome.
EPIDEMIOLOGY OF CRANIOSINOSTOSI
On the basis of the most recent statistics, it seems that a child suffers from craniosynostosis every 1800-3000 or so.
Regarding the most affected sex, several clinical studies have shown that 3 out of 4 patients are males. The reason why craniosynostosis is more widespread in the male population is completely unknown.
Craniosynostosis risk factors.
- Low birth weight
- Premature birth
- Advanced paternal age
- Maternal smoking during pregnancy
Symptoms and Complications
Most of the symptoms observed in the presence of craniosynostosis are due to an increase in pressure inside the skull . In medicine, the increase in pressure inside the skull is called intracranial hypertension or intracranial hypertension .
In the presence of craniosynostosis, intracranial hypertension is a consequence of the fact that the brain and other structures inside the skull do not have the right space to grow, so they go to push on the bony structures of the head.
Having said that, it is important to remember that, if the cranial sutures involved are many or if the condition is not treated in time, craniosynostosis can lead to a reduced development of cognitive faculties and a low IQ.
SYMPTOMS OF ENDOCRANIC HYPERTENSION
The possible symptoms of intracranial hypertension are:
- Persistent headache. Generally it gets worse in the morning and at night.
- Poor eyesight. They consist of double vision, blurred vision and blurred vision.
- He retched
- Irritability
- Puffy or prominent eyes
- Difficulty in following the movement of objects
- Hearing problems
- Respiratory problems
- Changes in mental status
- papilledema
The number of cranial sutures involved in the development of craniosynostosis has a decisive influence on the presence of intracranial hypertension.
For example, doctors have observed that the involvement of a single cranial suture induces intracranial hypertension in 15% of patients; while the involvement of at least two sutures leads to an increase in pressure inside the skull in at least 60% of patients.
In the presence of a mild form of craniosynostosis, intracranial hypertension begins to be problematic, causing the aforementioned symptomatology, around 4-8 years of life.
SIGNS OF THE CRANIOSINOSTOSI
Among the signs of craniosynostosis, the most common are:- Formations of rigid ridges along the cranial sutures
- Abnormalities of cranial fontanelles
- Head with dimensions not proportionate to the rest of the body
TYPES OF CRANIOSINOSTOSIS
The head shape of patients with craniosynostosis depends on which cranial sutures have closed prematurely.
Having observed this, doctors considered it appropriate to distinguish craniosynostosis in various types, depending on the cranial sutures involved.
The types of craniosynostosis are:
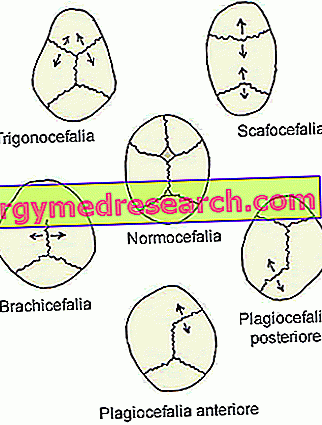
- Sagittal synostosis ( dolichocephaly or scaphocephaly ). It is the most common type of craniosynostosis; in fact, it characterizes about half of clinical cases.
Its presence coincides with the premature closure of the sagittal cranial sutures, located in the upper part of the skull, between the parietal bones.
From //en.wikipedia.org/wiki/Plagiocephaly
- Coronal craniosynostosis ( brachycephaly ) It is the second most common type of craniosynostosis; characterizes about one clinical case every four.
Its onset involves the premature fusion of the coronal sutures, which run between the frontal bone and the parietal bones.
- Metopic synostosis ( trigonocephaly ). It is a very rare type of craniosynostosis, which distinguishes only 4-10% of cases.
Its appearance coincides with the premature fusion of the metopic (or frontal) suture, which runs from the nose to the upper part of the head, separating the frontal bone in two. Generally, this suture naturally ossifies within the sixth year of life.
- The lambdoid synostosis ( plagiocephaly ). It is the rarest type of craniosynostosis. In fact, it is only 2-4% of clinical cases.
Its presence involves the early fusion of the lambdoid suture, located between the parietal bones and the occipital bone, in the back of the head.

COMPLICATIONS
In addition to impairing intellectual development, an untreated craniosynostosis can determine:
- The so-called obstructive sleep apnea syndrome .
- Permanent facial alterations, especially in the eyes and ears.
- Permanent deformities at the base of the skull (for example the malformation, or Arnold-Chiari syndrome).
The main cranial sutures involved in the craniosynostosis process. From the site: www.sciencebasedmedicine.org
- Hydrocephalus .

Diagnosis
To diagnose craniosynostosis, physical examination, evaluation of clinical history and radiological images provided by X-rays or CT scan to the head are essential.
If the craniosynostosis is of the syndromic type, it is also important to establish the morbid condition that led to its onset. Therefore, doctors could resort to blood tests and, above all, genetic counseling .
EXAMINATION OBJECTIVE
The physical examination consists of a careful analysis by the doctor of the clinical signs present on the subject's head, suspected of suffering from craniosynostosis.
In general, a pediatrician is responsible for carrying out this important diagnostic check.
CLINICAL HISTORY
The evaluation of clinical history is important for diagnostic purposes, as it includes questions related to the risk factors of craniosynostosis.
Therefore, the doctor (usually always a pediatrician) will investigate if:
- The baby is born premature or of low weight.
- What was the age of the father at the time of conception.
- If the mother smoked during pregnancy.
RADIOLOGICAL TESTS
X-rays and CT in the head serve more than anything to confirm the diagnosis and to show the doctor which cranial sutures have fused prematurely.
The knowledge of which cranial sutures are involved allows planning the most appropriate surgical treatment.
Treatment
Craniosynostosis is curable only by surgical intervention .
The latter consists of an operation of separation of the cranial sutures fusing early between them.
The final therapeutic goal of surgery is to provide the encephalic structures and some sense organs, such as the eyes, with that space needed to develop and function at its best.
BEST TIME TO INTERVENE
There is no total agreement, on the part of doctors, on what is the best time to perform craniosynostosis surgery.
According to some experts, the ideal period for the operation would be in late childhood, when the risk of a recurrence is lower (ie a second premature fusion of the cranial sutures). In case of relapse, in fact, the operation must be repeated and this is not recommended, given the delicacy of the procedure.
According to other experts, the most appropriate time would be in early childhood (between 6 and 12 months of life), when the skull is not yet completely ossified and the bones are still mouldable. The possibility of shaping bones (malleability) allows any morphological abnormalities of the bones to be resolved, which could cause serious aesthetic defects and functional problems (to the jaw or eyes) at a more mature age.
POSSIBLE SURGICAL APPROACHES
There are two different surgical approaches: traditional surgery, also called "open", and endoscopic surgery.
- Traditional (or "open") surgery .
It foresees general anesthesia (therefore the patient is unconscious during the entire operation) and the practice of a surgical incision on the head, exactly at the point where the radiological images showed the cranial anomaly.
Through the incision on the head, the operating surgeon (a neurosurgeon) removes the abnormal bone and entrusts it to a specialist in craniofacial surgery, who modifies it and gives it a shape that allows the normal development of the brain structures.
After the modification, the neurosurgeon reinserts the bone in its original position and closes the incision with stitches.
Like many traditional surgery, the "open" operation is somewhat invasive; however, to make it advantageous is the fact of being able to modify the bone structure precisely and with good results.
- Endoscopic surgery intervention .
It involves the use of an endoscope, a tool similar to a flexible tube, equipped with a fiber optic camera, at one end, and connected to a monitor.
From the operational point of view, it consists in inserting the endoscope into an opening made on the skull and in the separation, by means of the endoscope itself, of the fusesi suture precociously.
The neurosurgeon manages to orient himself inside the head, thanks to the images that the camera projects on the externally connected monitor.
The endoscopic surgery intervention is decidedly less invasive than the "open" operation (the hospitalization period is also shorter), however it has two drawbacks: it is indicated only for patients of a few months (6 in general), the which possess mouldable bones; is at greater risk of recurrence.
POST-OPERATIVE PHASE
Generally, a patient with craniosynostosis, subjected to surgery, must remain in the hospital for about 4-5 days after the operation. During this time, the neurosurgeon and his staff members periodically monitor their vital signs and check that everything is going well.
After the resignation, a series of periodic check-ups is scheduled, which are, at first, half-yearly and then, with the growth of the patient, annual.
Prognosis
The prognosis depends on various factors, including:
- The causes that induced craniosynostosis. Some genetic diseases responsible for this anomaly are very serious and have a poor prognosis.
- The position of the sutures merged early. If the sutures reside in positions that, for the neurosurgeon, are "uncomfortable" to reach, the craniosynostosis intervention becomes complicated and may not provide the desired results.


