What is Citrullinemia
Citrullinemia is a serious genetic disease with POSSIBLE PRENATAL DIAGNOSIS, of which two distinct forms are known: citrullinemia type 1 and citrullinemia type 2 ; in principle, citrullinemia determines:
- Excessive increase in blood citrulline
- Excessive increase in ammoniama and risk of coma (especially type 1)
- Increase in orotic acid (especially type 1)
- Arginine deficiency
- Impaired liver function
What is citrulline?
Citrulline (C 6 H 13 N 3 O 3 ) is a NON-essential amino acid that receives the name from the Latin noun of the first food from which it was isolated: the watermelon or citrullus .
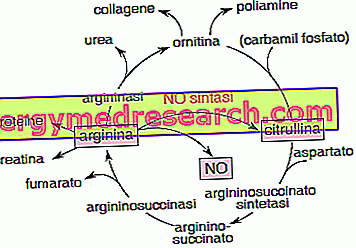
Both type 1 and type 2 citrullinemia are disorders related to alteration of the chromosomal heritage; however they are distinguished by the different etiopathogenesis and by the relative severity of symptoms and complications.
Citrullinemia type 1
Citrullinemia type 1 is an autosomal recessive genetic disorder; it causes an enzymatic deficiency of ARGININO-SUCCINICO SINTETASI (ASS), a fundamental component of the urea cycle. The disorder manifests itself in the first days of life with sudden and potentially inauspicious onset characterized by the HYPERAMMONEMIC COMA associated with LACTIC HYPERACIDOSIS. Sometimes the symptoms are later and less obvious but they show up with: anorexia, vomiting, hypotonia, growth retardation, psychomotor retardation and convulsions. The diagnosis of citrullinemia type 1 can be pre-natal performed by taking and analyzing amniotic fluid, or postnatal with:
- Hematochemistry: related to increased ammoniama
- Plasmatic and urinary amino acid chromatography: concerning the increase of citrulline, glutamine and alanine, and in parallel a reduction of arginine associated with orotic aciduria.
Citrullinemia type 2
Citrullinemia type 2 is a genetic disease very common in Japan caused by the mutation of the gene that translates for the enzyme DESOSSIRIBONUCLEASI 2 (AGC 2), a condition that also causes a deficiency of the mitochondrial transporter of aspartate / glutamate. Type 2 citrullinemia has a late onset, that is, in adulthood, and although of moderate form compared to type 1 citrullinemia, it is characterized by symptoms and complications that are anything but negligible, but are aggravated by other disorders or interaction with some drugs. Type 2 citrullinemia can cause: mild mental retardation, associated with liver failure requiring transplantation that is NOT always decisive.
The hypercitrullinemia food therapy is hypo-proteic assisted in supplementation with arginine, benzoate and sodium phenylbutyrate.


